

The urea cycle, also known as the ornithine cycle, is a crucial metabolic pathway in the liver that converts toxic ammonia into urea for excretion from the body. Ammonia is a byproduct of amino acid and protein metabolism, and its accumulation can be toxic, particularly to the central nervous system. The urea cycle ensures that ammonia is efficiently removed from the body. Disruptions in this cycle can lead to serious metabolic disorders known as urea cycle disorders (UCDs).
Urea Cycle: Overview
Location: The urea cycle occurs primarily in the liver, with steps distributed between the mitochondria and the cytoplasm of hepatocytes.
Key Steps and Enzymes
The urea cycle involves five main enzymatic steps:
1. Formation of Carbamoyl Phosphate:
Enzyme: Carbamoyl phosphate synthetase I (CPS1)
Location: Mitochondria
Reaction:
NH4+ + HCO3− + 2ATP → Carbamoyl phosphate + 2ADP + Pi
2. Formation of Citrulline:
Enzyme: Ornithine transcarbamylase (OTC)
Location: Mitochondria
Reaction:
Carbamoyl phosphate + Ornithine → Citrulline + Pi
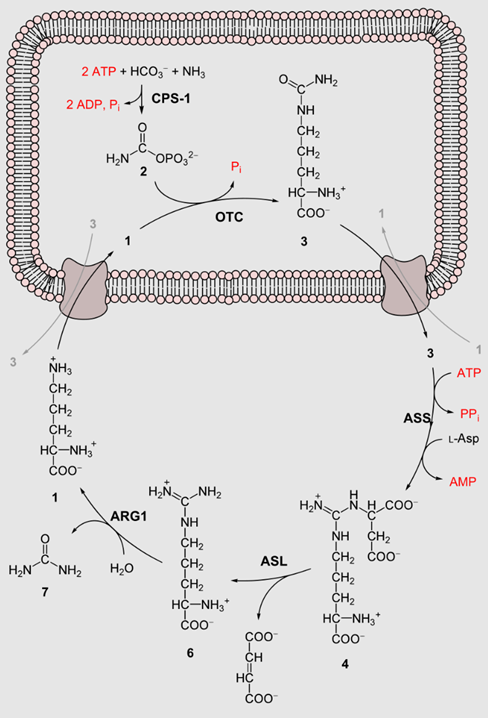
3. Formation of Argininosuccinate:
Enzyme: Argininosuccinate synthetase (ASS)
Location: Cytoplasm
Reaction:
Citrulline + Aspartate + ATP → Argininosuccinate + AMP + PPi
4. Formation of Arginine:
Enzyme: Argininosuccinate lyase (ASL)
Location: Cytoplasm
Reaction:
Argininosuccinate → Arginine + Fumarate
5. Formation of Urea:
Enzyme: Arginase
Location: Cytoplasm
Reaction:
Arginine + H2O → Ornithine + Urea
Overall Reaction:
NH4+ + HCO3− + Aspartate + 3ATP + H2O → Urea + Fumarate + 2ADP + 2Pi + AMP + PPi
Regulation of the Urea Cycle
The urea cycle is regulated by several factors to meet the body’s metabolic demands:
1. Allosteric Regulation:
N-Acetylglutamate (NAG): An essential allosteric activator of CPS1, synthesized from glutamate and acetyl-CoA by N-acetylglutamate synthase (NAGS).
2. Substrate Availability: The concentrations of ammonia, bicarbonate, and aspartate directly affect the flux through the urea cycle.
3. Hormonal Regulation: Glucagon and Cortisol: These hormones upregulate the expression of urea cycle enzymes during fasting and stress.
Urea Cycle Disorders (UCDs)
Urea cycle disorders are genetic conditions resulting from deficiencies in one of the enzymes or transporters involved in the urea cycle. These disorders lead to hyperammonemia (elevated ammonia levels in the blood), which can cause severe neurological damage and other systemic effects.
Common Urea Cycle Disorders:
1. Carbamoyl Phosphate Synthetase I Deficiency (CPS1 Deficiency):
Inheritance: Autosomal recessive
Symptoms: Lethargy, vomiting, seizures, and coma in neonates; hyperammonemia
Diagnosis: Elevated blood ammonia, low or undetectable citrulline
2. Ornithine Transcarbamylase Deficiency (OTC Deficiency):
Inheritance: X-linked recessive
Symptoms: Similar to CPS1 deficiency; can present later in life with episodes of hyperammonemia
Diagnosis: Elevated blood ammonia, low citrulline, elevated orotic acid in urine
3. Argininosuccinate Synthetase Deficiency (Citrullinemia Type I):
Inheritance: Autosomal recessive
Symptoms: Hyperammonemia, vomiting, lethargy, seizures, and mental retardation
Diagnosis: Elevated citrulline levels in blood
4. Argininosuccinate Lyase Deficiency (Argininosuccinic Aciduria):
Inheritance: Autosomal recessive
Symptoms: Hyperammonemia, developmental delay, brittle hair, and liver abnormalities
Diagnosis: Elevated argininosuccinate in blood and urine
5. Arginase Deficiency (Argininemia):
Inheritance: Autosomal recessive
Symptoms: Progressive spasticity, developmental delay, and episodic hyperammonemia
Diagnosis: Elevated arginine levels in blood
Management and Treatment of UCDs
The management of urea cycle disorders focuses on reducing ammonia levels and preventing hyperammonemia through dietary modifications, medications, and in severe cases, liver transplantation.
Dietary Management:
Protein Restriction: Limiting protein intake to reduce ammonia production.
Essential Amino Acids: Supplementing with essential amino acids to prevent deficiencies while controlling total protein intake.
Medications:
Ammonia Scavengers: Drugs like sodium phenylbutyrate and sodium benzoate help to remove excess ammonia by forming compounds that can be excreted in urine.
Arginine or Citrulline Supplements: Depending on the specific enzyme deficiency, supplementation can help to drive the urea cycle and reduce ammonia levels.
Emergency Treatment:
Hemodialysis: In cases of acute hyperammonemia, hemodialysis may be used to rapidly reduce blood ammonia levels.
Intravenous Glucose and Lipids: Providing alternative energy sources to reduce protein catabolism and ammonia production.
Liver Transplantation: Considered in severe cases where metabolic control is difficult to achieve with medical and dietary management. Transplantation can restore normal urea cycle function.
Summary
The urea cycle is a vital biochemical pathway that detoxifies ammonia by converting it to urea for excretion. Deficiencies in the enzymes or transporters of the urea cycle lead to urea cycle disorders, characterized by hyperammonemia and associated neurological and systemic effects. Effective management of UCDs requires a combination of dietary modifications, medications, and, in severe cases, liver transplantation to prevent hyperammonemia and its complications. Early diagnosis and treatment are critical to improving outcomes and quality of life for individuals with UCDs.