

Sickle cell anemia is a hereditary blood disorder characterized by the production of abnormal hemoglobin, known as hemoglobin S (HbS). This leads to the deformation of red blood cells into a sickle shape, which causes various complications including pain, anemia, infections, and organ damage. This note explores the genetic basis, pathophysiology, symptoms, diagnosis, treatment, and prevention of sickle cell anemia.
Genetic Basis
Sickle cell anemia is caused by a mutation in the HBB gene on chromosome 11, which encodes the beta-globin subunit of hemoglobin. This mutation results in the substitution of valine for glutamic acid at the sixth position of the beta-globin chain, forming hemoglobin S (HbS). The disease is inherited in an autosomal recessive pattern, meaning that an individual must inherit two copies of the sickle cell gene (one from each parent) to develop the disease. Individuals with only one copy of the sickle cell gene are carriers (sickle cell trait) and usually do not exhibit symptoms.
Pathophysiology of Sickle cell anemia
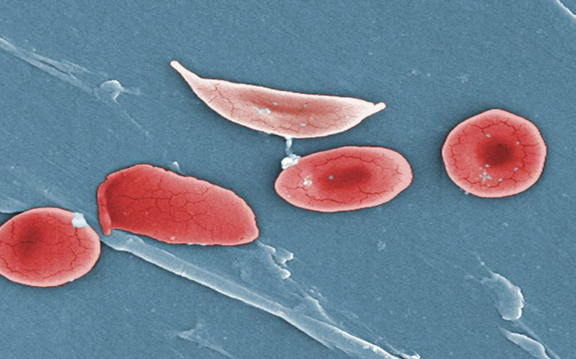
The primary pathophysiological feature of sickle cell anemia is the polymerization of deoxygenated HbS, which causes red blood cells to deform into a rigid, sickle shape. These sickle cells have several detrimental effects:
1. Decreased Flexibility: Sickle cells are less flexible than normal red blood cells and can obstruct small blood vessels, leading to vaso-occlusive crises.
2. Shortened Lifespan: Sickle cells have a shorter lifespan (10-20 days) compared to normal red blood cells (about 120 days), leading to chronic hemolytic anemia.
3. Hemolysis: The continuous destruction of sickle cells releases free hemoglobin, which can cause jaundice and gallstones.
4. Vascular Occlusion: Blocked blood vessels reduce blood flow and oxygen delivery to tissues, causing pain, inflammation, and tissue damage.
Symptoms of Sickle cell anemia
The symptoms of sickle cell anemia can vary in severity and include:
1. Anemia: Fatigue, weakness, and pallor due to chronic hemolytic anemia.
2. Pain Crises: Sudden episodes of severe pain, often in the bones, chest, abdomen, and joints (vaso-occlusive crises).
3. Swelling: Swelling of the hands and feet (dactylitis) in infants and young children.
4. Frequent Infections: Increased susceptibility to infections due to splenic dysfunction or autosplenectomy.
5. Delayed Growth and Puberty: Slowed growth and delayed puberty in children and adolescents.
6. Organ Damage: Chronic damage to organs such as the spleen, liver, kidneys, and lungs due to repeated episodes of vascular occlusion and hypoxia.
7. Acute Chest Syndrome: A life-threatening complication characterized by chest pain, fever, cough, and difficulty breathing, often triggered by infection, fat embolism, or pulmonary infarction.
Diagnosis of Sickle cell anemia
Diagnosis of sickle cell anemia involves a combination of family history, physical examination, and laboratory tests:
1. Newborn Screening: Routine screening for sickle cell disease is performed at birth in many countries.
2. Hemoglobin Electrophoresis: A laboratory test that separates different types of hemoglobin and confirms the presence of HbS.
3. Complete Blood Count (CBC): Indicates anemia and the presence of abnormal red blood cells.
4. Peripheral Blood Smear: Reveals sickle-shaped red blood cells.
5. Genetic Testing: Identifies mutations in the HBB gene.
Treatment of Sickle cell anemia
The treatment of sickle cell anemia focuses on managing symptoms, preventing complications, and improving quality of life:
1. Pain Management: Analgesics such as acetaminophen, NSAIDs, and opioids for severe pain crises. Hydration and heat application to affected areas.
2. Hydroxyurea: A medication that increases fetal hemoglobin (HbF) production, reducing the frequency of pain crises and acute chest syndrome.
3. Blood Transfusions: Used to treat severe anemia, acute chest syndrome, and to reduce stroke risk.
4. Bone Marrow/Stem Cell Transplant: The only potential cure, but limited by availability of suitable donors and associated risks.
5. Infection Prevention: Vaccinations and prophylactic antibiotics (e.g., penicillin) to prevent infections. Regular monitoring and early treatment of infections.
6. Folic Acid Supplementation: To support red blood cell production.
7. Lifestyle Modifications: Staying hydrated, avoiding extreme temperatures, and managing stress.
Prevention of Sickle cell anemia
Preventive measures for sickle cell anemia include:
1. Genetic Counseling: For individuals with a family history of sickle cell disease to understand their risk and reproductive options.
2. Prenatal Screening: Genetic testing during pregnancy to determine if the fetus has sickle cell disease or is a carrier.
3. Newborn Screening: Early detection and intervention to manage the disease and prevent complications.
Conclusion
Sickle cell anemia is a complex and serious genetic disorder with significant health implications. Understanding the genetic basis, pathophysiology, symptoms, diagnosis, treatment, and prevention of sickle cell anemia is crucial for effective management and improving patient outcomes. Advances in medical care and research continue to enhance the quality of life for individuals with sickle cell anemia, emphasizing the importance of early diagnosis, comprehensive care, and ongoing support.
 Join Our Telegram Channel
Join Our Telegram Channel